- 移动端


北京义翘神州科技股份有限公司(Sino Biological Inc.)品牌商
17 年
手机商铺
- NaN
- 0
- 0
- 2
- 2
北京义翘神州科技股份有限公司(Sino Biological Inc.)
入驻年限:17 年
- 联系人:
客服部
- 所在地区:
北京
- 业务范围:
技术服务、试剂、抗体、细胞库 / 细胞培养、ELISA 试剂盒
- 经营模式:
生产厂商
推荐产品




公司新闻/正文
「WB急救室」蛋白煮样不盲从,3连问洞悉变性背后的4大关键机制
663 人阅读发布时间:2026-03-24 17:22
在Western Blot(WB)实验中标准化流程中,有一步骤常常被忽视。
因为他太简单了,小case,不值一提
简单到只需放在水浴锅中煮上三五分钟就行
是的,就是煮样,蛋白质煮沸处理。
可你是否知道?
煮的好,条带清晰漂亮
煮不好,结果就是一团糟
这难道就是传说中的“卡脖子”技术?
接下来,咱们就一起来揭开煮样背后的“底层逻辑”吧!
一问:煮样的目的与原理是什么?
简单来说就是破坏蛋白质的三维结构,使蛋白质分子内部的氢键、二硫键断裂,成为线性多肽链。蛋白与SDS结合后带上负电荷。这样在电泳过程中,蛋白质从负极移动到正极,迁移率只受分子量大小的影响,与空间阻力、电荷性质无关,确保电泳结果的准确性。同时还能暴露更多的抗原表位,提高蛋白与抗体的结合效率,增强检测信号。
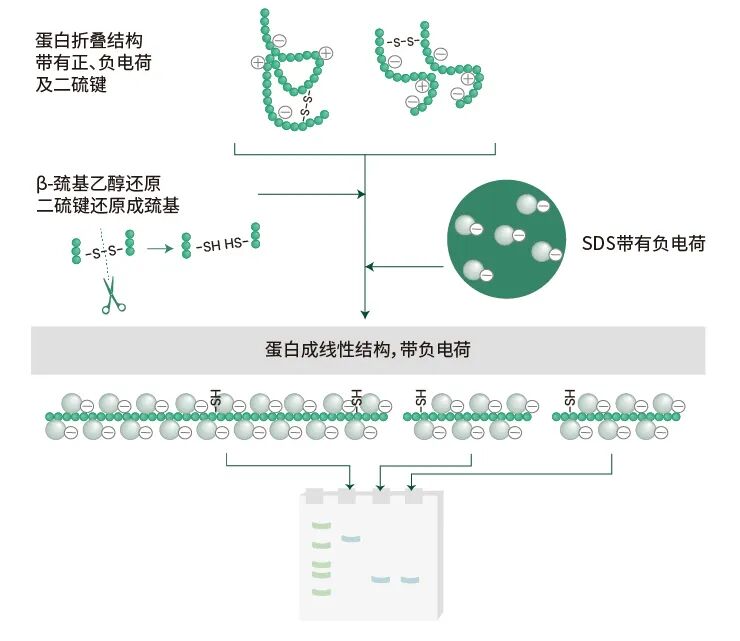
SDS-PAGE分离蛋白质的原理(蛋白变性前具有折叠结构。带正负电荷、二硫键,通过二硫键还原、结合SDS,使蛋白展开成线性结构,并带上与多肽长度成正比的负电荷,在电场中向正极移动)
通过高温(如95℃-100℃)和还原剂(如β-巯基乙醇或DTT)的共同作用,使蛋白质分子的氢键、疏水键、二硫键等断裂,使其二级、三级和四级结构展开为线性多肽链的一级结构。
蛋白质的高级结构可能成为WB实验中的“绊脚石”,对蛋白质的迁移率造成影响,导致条带模糊、位置异常等。煮样消除了蛋白质空间构象对电泳结果的干扰,确保蛋白质的迁移率仅与其分子量大小有关。
SDS(十二烷基硫酸钠)是一种阴离子去污剂,能与变性后的蛋白质按质量比约1.4:1结合,赋予所有蛋白质相似的电荷密度。SDS与蛋白质的结合并非自发,需要一定的物理化学条件,高温是最简便快捷的。煮沸处理能够打破蛋白质内部的非共价键,确保蛋白质的完全变性和解聚。
SDS的十二烷基链可以插入蛋白质的疏水核心区域。破坏蛋白质三级和四级结构间的疏水相关作用。强还原剂β-巯基乙醇或DTT断裂半胱氨酸残基间的二硫键,进一步破坏蛋白质的四级结构。SDS还能断裂蛋白质分子内和分子间的氢键,破坏蛋白分子的二级和三级结构,使蛋白质去折叠。SDS与蛋白质的疏水部分结合后,在蛋白表面形成一层负电荷(如图所示)。总之,SDS通过破坏蛋白质的疏水作用、氢键、二硫键,并形成带有大量负电荷的SDS-蛋白质复合物,使蛋白质在电泳过程中以一级线性结构进行分离。
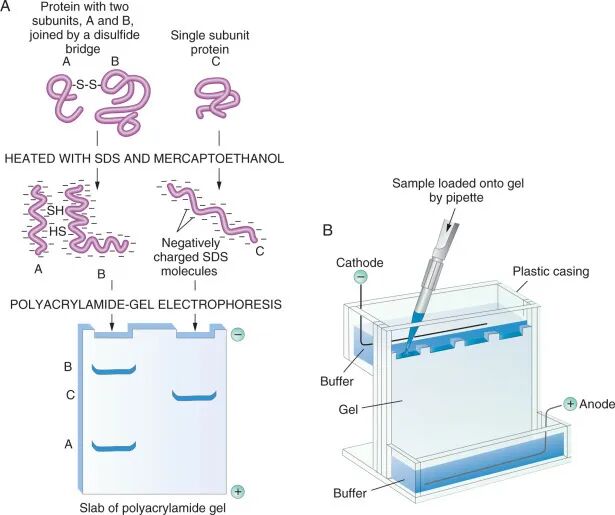
Modified from Alberts B et al., Molecular Biology of the Cell, 4th ed. New York, NY: Garland Science, 2002.
煮沸处理能够淬灭蛋白样品中存在的一些蛋白酶,防止样品降解,确定蛋白质在电泳过程中保持完整。蛋白酶是一类能够水解蛋白质的酶类,如半胱氨酸蛋白酶、天冬氨酸蛋白酶、丝氨酸蛋白酶、金属蛋白酶等。它们可能会随时出现在样品中,一旦被激活,就会像“剪刀”一样,将蛋白质分解成碎片,造成电泳出现多条带,使得WB结果非特异性条带增加或目的条带变弱甚至“白板”。因此需要及时灭活样品中的蛋白酶。煮沸处理是一种有效的方法,能够确保蛋白质在电泳过程中的完整性。
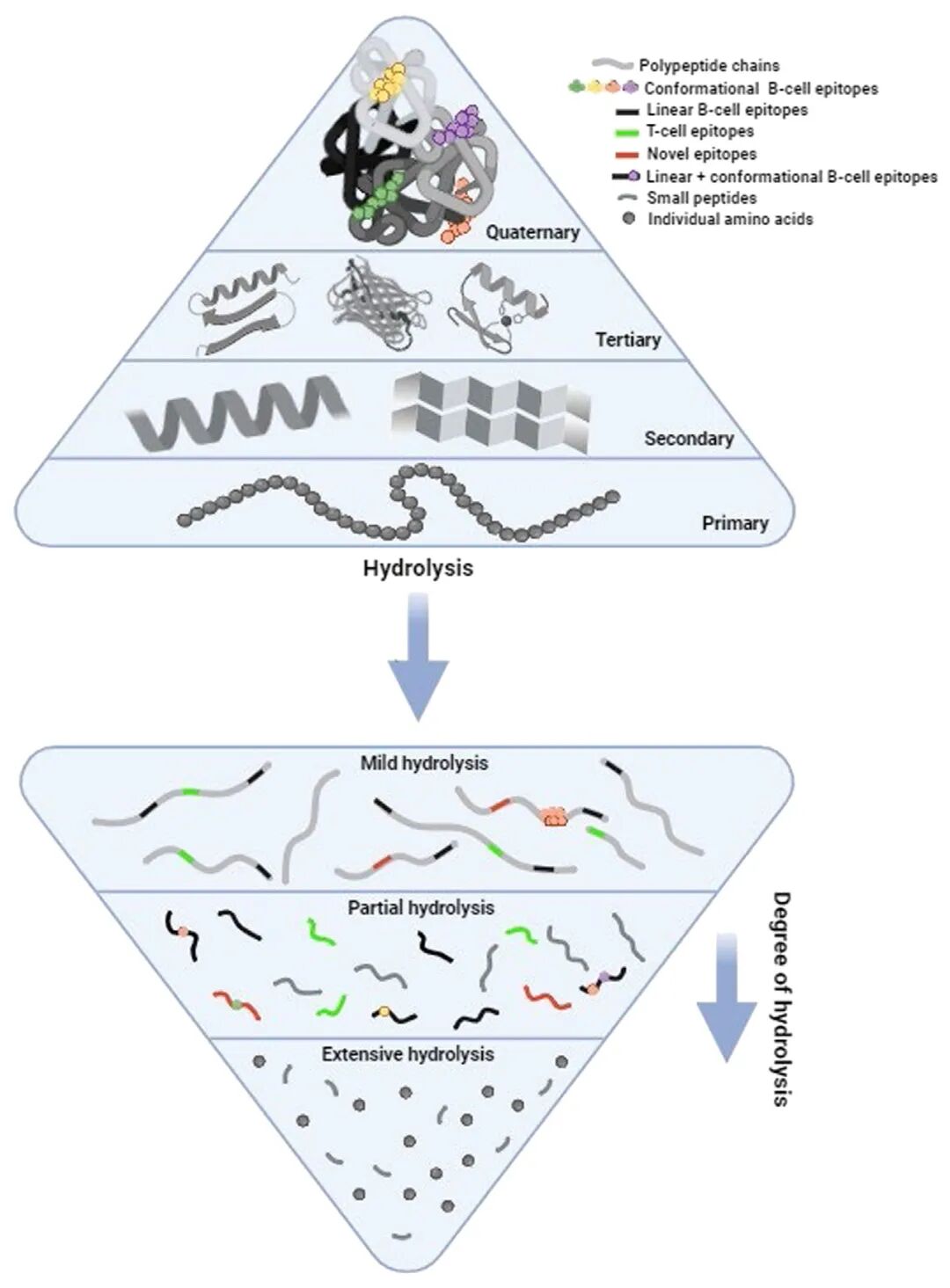
源自书籍:Hydrolysis in food processing and its impact on allergenicity of food
95-100℃使得蛋白质分子内部相互作用力减弱,不易发生聚集和沉淀。此外,煮沸还能够去除样品中的气泡或其它难溶杂质。每次上样前进行煮样,能够确保实验条件一致,提高WB结果的可靠性和重复性。蛋白煮样不充分可能会导致条带模糊或缺失,如果样品还存在高级结构会影响电泳迁移率,导致条带位置不准确、拖尾、弥散等。
二问:上样缓冲液中各组分的作用是什么?
上样缓冲液,即loading buffer,源自Ulrich K.Laemmli教授发明的Laemmli缓冲液。“祖宗级”配方:
The samples (0.2-0.3 ml.) contained the final concentrations (" final sample buffer"): 0.0625 M Tris--HCl (pH 6.8), 2 percent SDS, 10 percent glycerol, 5 percent 2-mercaptoethanol and 0.001 percent bromophenol blue as the dye.
——Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature, 1970.
loading buffer的详细组分可能在不同的实验室会有细微差异,比如2X、4X、5X等,但其主要组分是一样的,包括:
如上,常用的变性剂是SDS,能够破坏维持蛋白质高级结构的作用力,包裹在蛋白质一级结构表面,使其带负电荷。除此之外还可以用LDS(十二烷基硫酸锂)。LDS去污能力与SDS相当,适合低温环境(4-25℃)。
通常1X上样缓冲液中含有2-5%(w/v)的SDS或LDS。SDS常用于Tris-甘氨酸胶体系,LDS用于Bis-Tris和Tris-乙酸胶体系。
常见的还原剂有二硫苏糖醇(DTT)和β-巯基乙醇(2-ME),用于破坏蛋白质间的二硫键,并防止半胱氨酸的氧化,与SDS协同作用,使蛋白质线性化。两者在蛋白样品制备中的还原效果相差不大。
通常1X上样缓冲液中含有1%(w/v)的还原剂。需要注意的是DTT易被氧化,2-ME易挥发,二者都不稳定,最好现用现加。
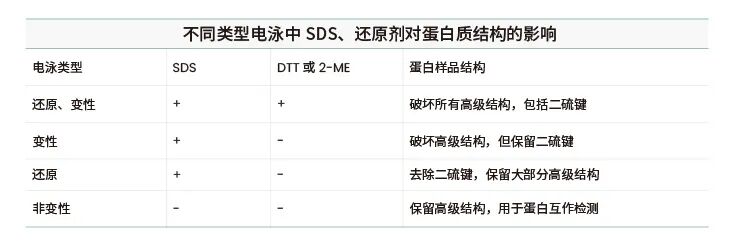
上样缓冲液常用Tris-HCl体系,pH保持在6.8左右,防止低温保存过程中蛋白质的肽键断裂,还可以抑制酶促反应,保证蛋白质的稳定。
增加样品密度,确保样品下沉到点样孔,防止样品飘散。通常1X缓冲液中浓度在5-10%(v/v)之间,小分子蛋白样品可适当调整到10-15%。
用于指示样品在凝胶中的位置。常用的示踪染料除了溴酚蓝还有考马斯亮蓝G-250、酚红等。一般终浓度为0.1%(w/v)。

三问:怎么煮样才科学?
蛋白样品在完成提取后应尽快进行煮沸处理,防止样品中可能存在的蛋白酶“作妖”,导致样品降解。
常规煮沸处理:加入上样缓冲液后,可以用移液器轻轻吹吸几次或者涡旋振荡3-5s,充分混匀样品和缓冲液,保证受热均匀。然后放入沸水浴或PCR仪中,95℃-100℃煮沸5-10分钟。煮样完成后,迅速以12000rpm高速离心3-5分钟。离心后样品置于冰上,准备上样电泳。
煮样时间可根据蛋白浓度、分子大小、上样缓冲液比例等因素进行调整。如蛋白浓度高,可适当增加煮样时间,但最好不超过10min。如果10min后仍然吸不出样品,看增加上样缓冲液稀释或对样品进行超声,再次煮样5-10min看看效果。若煮样时间过长,样品出现明显的蛋白沉淀和水分层,建议丢弃吧。大分子蛋白(如IgM 220kd)可适当延长时间。
煮样前需要按照一定比例加入loading buffer,主要与buffer浓度有关。以2X buffer为例,按1:1的体积比在样品中加入buffer,如10μL样品需加入10μL 2X buffer,制成20μL 混合液。5X buffer则需按4:1的体积比加入buffer,即10μL样品需加入2.5μL 5X buffer,制成12.5μL 混合液。
前提条件是蛋白样品的浓度是一样的。如果不一样则需要加入不同体积的buffer。如表格所示已测定4种样品的蛋白浓度。为了保证蛋白质上样量一致,如每个泳道的上样量为30μg,则每个样品的上样量分别为:3.99μL、5.74μL、4.61μL、2.48μL。
|
|
样品1 |
样品2 |
样品3 | 样品4 |
|
蛋白浓度(μg/μL) |
9.41 |
6.54 |
8.12 | 15.14 |
|
蛋白体积(μL) |
3.19 |
4.59 |
3.69 | 1.98 |
|
5x loading buffer体积(μL) |
0.8 |
1.15 |
0.92 | 0.5 |
|
电泳上样量(μL) |
3.99 |
5.74 |
4.61 |
2.48 |
用蛋白裂解液或超纯水将样品稀释到相同的浓度更利于电泳操作。如上样品,可以按照下表先统一稀释到5μg/μL(以100μL为例)。稀释后的样品可分装为10μL,每份加入2.5μL 5× buffer。
|
|
样品1 |
样品2 |
样品3 | 样品4 |
|
蛋白浓度(μg/μL) |
9.41 |
6.54 | 8.12 | 15.14 |
|
蛋白原液体积(μL) |
53.13 |
76.45 |
61.58 | 33.03 |
|
裂解液或超纯水体积(μL) |
46.87 |
23.55 |
38.42 | 66.97 |